Research Details
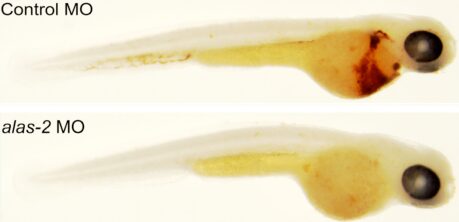
Sideroblastic anemias are acquired or inherited anemias that result in a decreased ability to synthesize hemoglobin in red blood cells and result in the presence of iron deposits in the mitochondria of red blood cell precursors. A common subtype of congenital sideroblastic anemia is due to autosomal recessive mutations in the SLC25A38 gene. The current treatment for SLC25A38 congenital sideroblastic anemia is chronic blood transfusion coupled with iron chelation. The function of SLC25A38 is not known. Here we report that the SLC25A38 protein, and its yeast homolog Hem25, are mitochondrial glycine transporters required for the initiation of heme synthesis. To do so, we took advantage of the fact that mitochondrial glycine has several roles beyond the synthesis of heme, including the synthesis of folate derivatives through the glycine cleavage system. The data were consistent with Hem25 not being the sole mitochondrial glycine importer, and we identify a second SLC25 family member Ymc1, as a potential secondary mitochondrial glycine importer. Based on these findings, we observed that high levels of exogenous glycine, or 5-aminolevulinic acid (5-Ala) a metabolite downstream of Hem25 in heme biosynthetic pathway, were able to restore heme levels to normal in yeast cells lacking Hem25 function. While neither glycine nor 5-Ala could ameliorate SLC25A38 congenital sideroblastic anemia in a zebrafish model, we determined that the addition of folate with glycine was able to restore hemoglobin levels. This difference is likely due to the fact that yeast can synthesize folate, whereas in zebrafish folate is an essential vitamin that must be obtained exogenously. Given the tolerability of glycine and folate in humans, this study points to a potential novel treatment for SLC25A38 congenital sideroblastic anemia.